Introduction
It’s been a busy past few months at Open Insulin! We’ve had significant progress on all fronts, especially in moving the engineering of our organisms forward towards the point where production pilots can be started in collaboration with external partners. Details below.
Wet Lab Progress Overview
Our overall goal with this grant is to produce a fast-acting insulin (lispro) and a slow-acting insulin (glargine) at concentrations of 0.25-0.5 g/L and 1g/L, respectively.
The steps in the production of insulin are as follows:
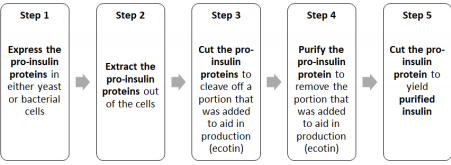
To accomplish our overall goal, we have 5 scientific objectives:
1. Molecular Characterization (shape and structure) of the insulin analogs lispro and glargine that we produce
2. Establishing master cell banks (MCB) of cells that express lispro, glargine, and KEX2
3. Develop a purification process for the insulin analog lispro
4. Optimize the enzymatic reaction to convert proinsulin to insulin
5. Scale-up for expression and purification of the proteins
Our results so far are that we have:
● Established a successful collaboration with the Molecular Characterization and Analysis Complex (MCAC) at UMBC for characterization of lispro samples (Objective 1)
● Confirmed that the proinsulin form of lispro is being expressed in bacterial cells (Objective 1)
● Have progressed our strategy for purifying the protein from cells (a new osmotic shock method) and for transforming the protein into proinsulin (Objective 3)
● Acquired the specialized equipment to scale-up growth of cells (a bioreactor; Objective 5) and to identify cells that produce the highest levels of insulin (a pipette robot; Objective 2)
● Used the specialized equipment (pipette robot) to begin developing methods for identifying cells that produce the highest levels of insulin (Objective 2)
● Launched the 2nd Open Insulin summer internship program for high school and college students including, for the first time, summer research experiences at BUGSS
● Identified a company that is our first candidate as a contract manufacturing organization for the next stage of scaling-up insulin production
● The project and was featured in Technical.ly Baltimore (Baltimore Under Ground Science Space, foundation receive $137K grant to make affordable insulin) and we did a segment for WYPR’s “On the Record with Sheilah Kast.”
Our immediate next steps are:
● Optimize growth of cells in the new bioreactor to scale up production of proinsulin (Objective 5)
● Optimizing the procedure for purifying the protein from cells (the osmotic shock method) and confirming that it is the best option for purification (Objective 3)
● Further advance methods for transforming the protein into proinsulin (Objective 3), purifying, and characterizing that protein (Objective 1) using a new analytical column (HPLC) that should have better resolution
● Finalize method for identifying cells that produce the highest levels of insulin with the specialized equipment (pipette robot; Objective 2)
DETAILED REPORT
Aim 1: Molecular Characterization of insulin analogs Lispro and Glargine
We need to be able to characterize the two forms of insulin that we are making (short-acting lispro and long-acting glargine) to make sure that the insulin protein has the correct sequence, that it has the proper shape (which is crucial for it to function properly), and to ensure that there are no other contaminating forms of insulin (or other proteins) present. This information will also give us an indication about the purity and the activity of the insulins, and we can use all of this information to optimize the purification process.
What we have achieved so far:
1. We have collaborated with the Molecular Characterization and Analysis Complex (MCAC) at the University of Maryland Baltimore County for experiments to determine whether proinsulin and insulin are in the correct shape and chemical form (ie, has the appropriate disulfide bridges and post-translational modifications). These results indicate that we are able to make the proinsulin form of lispro.
● We received different results when we repeated the experiment with a slightly different form of the protein (reduced versus non reduced), which indicates that a structure has formed in the protein called a disulfide bridge.
● We are now well versed with this new protocol of mass spectrometry and can analyze results directly from the software (PEAKS).
2. We have made progress in using a second technique (HPLC) to verify the shape and identity of the insulin protein, and have now identified the appropriate HPLC column that will allow us to get the needed resolution to properly separate insulin and the other protein.
3. We have made progress on using a third technique (antibody detection) to verify the shape and identity of the protein. Led by the Oakland Open Insulin team, we have identified antibodies that bind to commercial forms of the insulin glargine. Since antibodies bind and recognize shape, binding with antibodies is a good indication of a protein having the right shape.
Aim 2: Establishing a master cell bank (MCB) for
● Pichia pastoris clone that produces insulin Glargine with a high yield (~1 g/L) ● Pichia pastoris clone that produces ssKEX2 enzyme with a high yield (~1 g/L)
● Escherichia coli clone that produces insulin Lispro-ecotin with a high yield (~0.25-0.5 g/L)

Identifying high expressing yeast cells (clones) can take thousands of trials. To achieve this level of screening, we obtained a pipetting robot (an Opentrons robot) that we plan to transform into a fully automated system that can test thousands of clones per week and help us to automate screening.
The robot will aid us with multiple steps in the process of finding cells that produce the highest levels of protein production:
● Selecting and isolating individual cells
● Placing the individual cells in growth media so they can grow and produce proinsulin protein
● Evaluating the amount of protein produced by the cells to identify cell clones with the highest protein expression levels
What we have achieved so far:
1. We have confirmed that having the robot place the cells in growth media (growing them 96 at a time in deep-well culture vessels) achieves similar growth as doing it manually (they achieve similar absorbance (OD=2-3) as in a flask), demonstrating that this set up is suitable for cell culture tests.
2. We confirmed the feasibility of screening for protein levels with the color-based BCA assay. A protocol was designed with the robot and tested.
a. One major concern is the fact that the assay is time sensitive, and that there will be an artifact in the measurement due to the time the robot takes to prepare the samples but we have ordered a multi-channel pipette to speed up the experiments.
b. Another issue that we may encounter is to adequately estimate the approximate concentration of protein in the growth media. In addition to our protein of interest, we will get a mixture of other proteins that have been secreted by the cells and the concentration of these proteins may vary. We will have to run multiple tests to figure out a BCA standard that can be used routinely to account for this effect.
3. We obtained a Pichia pastoris yeast clone that produces Glargine insulin, although the insulin is fused with a fluorescent protein, mTurquoise (described below).
Scientific summary of the work done on the fusion protein insulin Glargine and mTurquoise We modified the insulin glargine protein to add a fluorescent protein, mTurquoise. This is a common technique because it is easy to follow the fluorescence in the sample, which should reflect the amount of insulin protein and we can then easily determine the quantity of protein secreted. The amount of insulin protein will be estimated by the fluorescence level, and the amount of total protein will be determined with a color-based BCA assay. We hope that there will be a direct correlation between the amount of total protein and the amount of fluorescent protein. If this is the case, it means that the BCA assay will be a reliable tool since the natural variation in protein levels will be negligible.
The fluorescent protein mTurquoise was chosen because of the high fluorescence it displays at low pH, which is the condition necessary for expression of the proinsulin glargine. In our first experiments, none of the cells expressed the fluorescent protein. We realized that the pH of the solution had become really low (around pH 2.3). This extremely low pH could have affected either the production of the protein or the fluorescence of the protein (which is known to be affected by really low or high pH levels). When we added a buffer to the growth medium (a potassium phosphate buffer to maintain the pH at 5.8). Recent results show that some protein with a blue color is expressed once the pH is controlled. (Figure 1). We need to reproduce this results and we need to perform mass spectrometry analysis to confirm the presence of glargine attached to the mTurquoise fluorescent protein, but this is encouraging preliminary evidence that we may have achieved production of the glargine pro-insulin analog.
Figure 1: Concentrated supernatant from 3 different cultures of GS115 mTurquoise-Glargine, in 20mL, with an induction of 0.5% methanol during 2 days.
Next steps: Further alter the DNA and protein sequence to increase the protein yield
The proinsulin protein is produced by adding DNA (the gene for insulin) into cells and having cells produce the insulin protein. In addition to screening cells to see which one is most efficient at producing the insulin protein from the DNA, we can also employ a more design-oriented solution. Specifically, for glargine insulin two research papers describe modifications to the DNA sequence that lead to either increased production of the proinsulin protein (by adding a sequence linker between the insulin glargine and the mat alpha factor) or increased secretion of the insulin protein from the cell into the growth media (by suppressing the amino acid at the position 57–70). Both those solutions can really improve the level of expression. We have designed these experiments and will perform the molecular cloning experiments to make those alterations to the DNA and see the effect on protein production and secretion.
Aim 3: Developing a purification process for the insulin analog lispro

To produce lispro, another protein called ecotin has been fused to the proinsulin form of the protein. This is a common technique to enhance protein production and make sure that the protein goes to the correct location in the cell. There is evidence that the ecotin and the proinsulin have formed inappropriate chemical bonds (mismatched disulfide bridges). This is most likely the consequence of isolating the protein from the cell’s cytoplasm, which is not the optimal location for correct formation of disulphide bridges. Therefore, we are instead isolating the ecotin-proinsulin protein from a different region of the cell (the periplasm) which is a more suitable location for correct disulphide bridge formation. Amelia, an Open Insulin scientist working in Oakland, has developed a method (called osmotic shock) for isolating lispro from the cell’s periplasm. We have further been able to confirm that we are producing the correct protein using the techniques of Western blot and have been able to purify the extract using FPLC.
However, the low amount of protein that is present in the periplasm requires the use of bioreactors to scale up production. These efforts to grow cells in bioreactors are ongoing, but we have high expectations that when the bioreactor will be up and running, this method of isolating the protein from the periplasm instead of the cytoplasm will alleviate most of our issue. In the next few weeks, we plan to optimize the cell culture conditions in the bioreactor and test tise protocol for extracting proteins from the periplasm.
Aim 4: Optimization of the enzymatic reaction to convert proinsulin to insulin
The transformation of lispro from the ecotin-pro insulin form into the insulin form includes 3 steps:
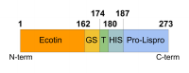
- Cleavage of ecotin from proinsulin (with thrombin protease) to produce proinsulin
- Purification to remove ecotin
- Transformation of the proinsulin into insulin (with trypsin)
Step 1: Cleavage of ecotin from proinsulin (with thrombin protease) to produce proinsulin
To obtain our final product, lispro insulin, we need to separate the proinsulin from ecotin. It needs to be cleaved to remove the ecotin sequence that was attached to help in proper formation of the protein. For this, we use the protease thrombin that can cleave the two apart. With 10 units of thrombin/mg and an incubation overnight, we can see that the large band (dark line in the figure) corresponding to the ecotin-lispro fusion is reduced, as expected, and two bands appear, each corresponding to the now separated ecotin and lispro (column 3, figure 2). This result confirms that we have successfully completed step 1 (cleavage of ecotin from proinsulin (with thrombin protease) to produce proinsulin).
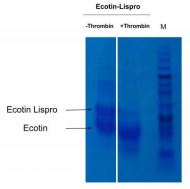
Figure 2: Test of digestion of the ecotin lispro with Thrombin protease observed by SDS PAGE. 10 unit of Thrombin per mg of protein was added. M corresponds to the protein ladder. When done at CCL by David A., with a different type of gel, the gel revealed that before cleavage there was a single protein size at 28kDa, representing the full length construct. Then after cleavage the gel showed two proteins sized at 17.5kDa and 10.5kDa. The larger of these two representing ecotin and the smaller representing proinsulin, which is the precursor to Lispro.
Step 2: Purification to remove ecotin
Ecotin has been shown to reduce the activity of enzymes, such as the one that we need to use for step 3. We therefore need to remove it from our sample before moving to the next step. When performing a common purification procedure (using a histidine tag) to remove the ecotin from the proinsulin, we surprisingly found that not all of the ecotin was removed (a lot of ecotin protein was observed in the elution fraction). We performed different experiments to try to further remove the ecotin:
● Added an additional purification step using ultracentrifugation with no success
● Tried to remove any ecotin that might be interacting with the proinsulin in a nonspecific way by increasing the amount of salt (up to 1M NaCl) after the digestion and incubating for 1h before the chromatography. This change reduced the amount of ecotin but still didn’t completely remove it (Figure 3).

Figure 3: SDS PAGE of sample used for the reverse chromatography experiments. The letters correspond to the different sample in the figure 4, A – Ecotin Lispro, B – Flow through of second affinity chromatography, C- Wash with 50mM TrisHCl pH 7.5, 40mM Imidazole, D – Elution with 50mM TrisHCl pH7.5, 300mM Imidazole
Since disturbing non-specific interactions between the ecotin and the proinsulin seems not to be enough, we formulated the hypothesis that ecotin and proinsulin lispro were linked by stronger bonds called disulfide bridges. Indeed, we see proteins that are larger than those expected which could correspond to ecotin and lispro attached together by some disulfide bridges (Figure 4). To test the hypothesis that ecotin and proinsulin are joined by these bonds, we treated the proteins with a chemical called DTT that would remove these bonds if they were present, and we see these larger protein forms reduced (Figure 3). This method is not specific to ecotin and lispro, so it is not excluded that those bands are not ecotin and lispro protein, but it is likely that they are. These results strongly suggest that we are having trouble removing the ecotin because it is inappropriately forming bonds with the proinsulin that prevent it from being removed.
Figure 4: Difference of visualisation of the ecotin lispro by SDS PAGE with and without reductive agent. Ecotin-lispro +DTT correspond to a sample of ecotin lispro with a loading solution with DTT, Ecotin-Lispro -DTT correspond to a sample of ecotin lispro with a loading solution without DTT. Because we could not remove all of the ecotin, we wanted to see whether the ecotin that remains would actually be a problem. Because the ecotin and proinsulin lispro were linked via disulfide bonds, it is possible that the ecotin that remains is actually nonfunctional and may not interfere with subsequent steps.

Therefore ran an experiment to confirm that the ecotin still present in the sample was indeed active and that it would inhibit the protease reaction (Step 3). We performed step 3 by treating the protein with the enzyme trypsin and then analyzed the results with chromatography. The protein that was not treated and the one that was treated with trypsin were similar, indicating that the trypsin was not able to cut the protein (figure 5).
Therefore, the remaining ecotin seems to be a problem that we need to find another solution to solve (see below).

Figure 4: Reverse chromatography with C18 column of sample from the second affinity chromatography and with a test of digestion with trypsin of the lispro insulin.
There is evidence that ecotin and proinsulin are joined by bonds (called disulphide bridges) that have formed inappropriately. This is a problem because if the ecotin is not properly cleaved in step 1, then it cannot be removed in step 2. Ecotin is known to inhibit the activity of the trypsin protein used in step 3, so if step 1 does not occur properly and the ecotin is not removed, then it is likely that step 3 will not occur properly either, as we have demonstrated above.
As described above, we think that the incorrect bond formation is most likely the consequence of isolating the protein from the cytoplasm which is not optimal for correct formation of disulphide bridges. We are pursuing the isolation of ecotin-proinsulin from the periplasm which is more suitable for correct disulphide bridge formation. However the low amount of protein secreted to the periplasm requires the use of bioreactors to scale up production. These efforts are ongoing.
We are also trying to overcome this issue in another way, by using a different enzyme in step 3 to transform the proinsulin into insulin; we believe that this enzyme might not be inhibited if the ecotin cannot be fully removed. We are already using this enzyme, ssKEX2, in the insulin glargine purification process, and we plan to run additional experiments to see whether it would be inhibited by the ecotin. Although ecotin inhibits many enzymes, a publication from 1983 indicates that ecotin will not inhibit the ssKEX2 protein. Our initial attempts using ssKEX2 that we made ourselves were unsuccessful. We have now purchased commercial ssKEX2 to test this hypothesis. If successful, this would allow us to complete step 3 and produce the final form of insulin even if the first step cannot be improved any further.
Aim 5: Scale-up for expression and purification for bioreactor with E.coli
The scale-up process is a crucial step towards cGMP manufacturing (Current Good Manufacturing Practice regulations enforced by the FDA) that can be done in house or outsourced to a Contract Manufacturing Organization (CMO). Relying on the experience and expertise of a CMO could be an advantage as well as increasing the rapidity of execution. We are progressing on building a working relationship with a CMO (see below), and this will take time and will require us to audit and establish detailed expectations. Because the processes developed for small bioreactors are similar to those of the middle size bioreactors (100-1000 L) used in CMO facilities, we are using small bioreactors in our facility to establish a robust process which can then be reproduced in a CMO facility to produce cGMP injectable insulin. The experience we gain using a bioreactor will help us to audit and select a suitable CMO partner as well as to continue our work developing a protocol for small scale manufacturing in facilities we establish directly within our own network.
After researching equipment and doing price assessment, we purchased two bioreactors (model Sartorius Biostat A) which come with sensors, computers, software and two vessels of 5L and 1L. Initial tests have been successful, and we will start by optimizing cell culture conditions to produce the proinsulin lispro. With these reactors to grow higher numbers of cells, we will be able to extract the ecotin-proinsulin protein from the periplasm using the osmotic shock method that we have adapted. We will then characterize the protein, but we believe that this new approach will solve our problem of incorrect bond formation (disulphide bridges) and the resulting problems that stem from it as described above.

Based on published work, we expect a yield around 50 mg/L. In collaboration with the business team, we estimate that the goal should be between 500 mg/L to 1 g/L to have a competitive cost of production. To improve the yield, we did an extensive bibliography research for producing this type of protein. One factor that can have a large effect on yield is the portion of the protein that enables the protein to be secreted into the periplasm compartment of the cell. We will therefore make four different versions of this portion of the protein and assess the impact on the yield of proinsulin.
Regulatory Update
Manufacturing
The team has been researching potential contract development and manufacturing organizations (CDMOs) based in Baltimore and the greater Maryland area. CDMOs are companies that you can contract to assist with the pharmaceutical development and manufacturing. This is a very standard practice in the industry.
We identified several organizations that are headquartered or operate in Maryland.
Post Market Surveillance
The Open Insulin Legal team has made progress in exploring how the requirement for post market surveillance may be fulfilled when considering the various manufacturing and distribution routes that are being considered. Post Marketing Surveillance is a requirement for market biologic drug products as outlined in the Code of Federal Regulations (CFR) Part 600 and entails a systemic approach to the collection and analysis of complaints, product reports, and adverse events. Feedback from this surveillance is used to address issues that may occur, both technical and medical, once the product is in the field. This can include investigations into reported events, risk assessments, review of labeling adequacy, and product enhancements.
Summer Internship Program
Applications are currently open for the Open Insulin Foundation Summer Internship Program. This is the second year that Open Insulin will be hosting a summer internship program that is open to high school and college students. The internship program allows students the ability to participate part time by working in teams under a mentor for 9 weeks. Two groups of up to 4 students each from the Baltimore area will be accepted as interns and will work under the leadership of 4 mentors to participate in lab work to further the goals of this grant. Students will help with some of the routine screening of cells to find those that produce the highest level of protein and will help make further modifications to the insulin protein in the hope that some of these side projects might turn out to be fruitful. This internship opportunity includes an educational curriculum in addition to the team projects. Last year’s Open Insulin internship program had 20 students in 5 mentorship teams and was beneficial both for the Open Insulin project in terms of the work produced and for the interns who reported that they learned invaluable life skills. Already applications to participate have been very strong, and we are enthused to expand the number of Baltimore participants in the Open Insulin initiative. We plan to reach out to local media again, especially television media, to let them know about the internship program and to invite them to film the students and feature a story on their broadcasts.